Western blot je metoda používaná pro kvalitativní nebo semikvantitativní detekci určitého proteinu ve vzorku. Metoda je tvořena třemi základními kroky: 1. elektroforetickou separací proteinů, 2. přenosem separovaných proteinů, 3. detekcí proteinů.
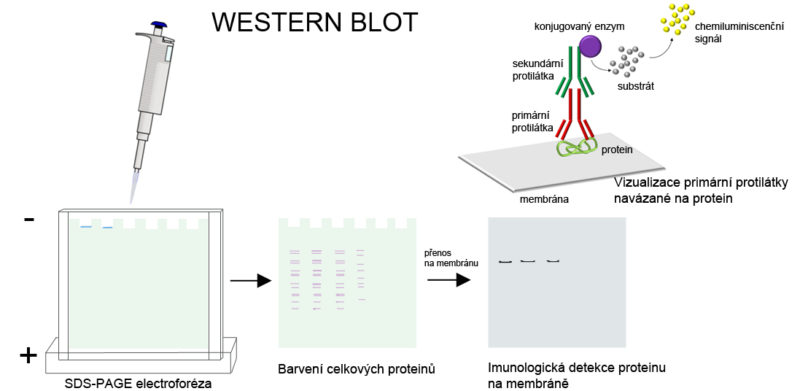
• Pro separaci proteinů se nejčastěji využívá gelová elektroforáza, nejčastěji je to SDS-PAGE (SDS polyacrylamide gel electrophoresis). Vlivem SDS jsou proteiny denaturovány a proteiny získávají záporný náboj a cestují tak v elektrickém poli od záporného pólu ke kladnému. Při elektroforéze jsou proteiny děleny podle své hmotnosti (udává se v kDa). Malé proteiny cestují akrylamidovým gelem rychleji, proteiny o velké molekulové hmotnosti se pohybují směrem k anodě pomaleji. Někdy je využívaná nativní-PAGE, kdy jsou proteiny separovány na základě svého náboje.
• Separované proteiny jsou přeneseny na membránu. Přítomnost daného proteinu je na membráně detekována pomocí protilátky, tzv. primární protilátky proti tomuto proteinu. Primární protilátka se navazuje na protein a v dalším kroku je rozpoznávaná sekundární protilátkou.
• Sekundární protilátka se tedy váže na primární protilátku. Přítomnost sekundární protilátky je detekována fluorescenčně či chemiluminiscenčně, a to v závislosti na značení, které sekundární protilátka nese.
• Velikost signálu je vyhodnocována srovnáním s naneseným proteinovým markerem, což je komerčně dostupná směs proteinů o známé velikosti.
————————————————————————————————————————————————————————————–
PŘÍPRAVA VZORKŮ
Lyzační pufry. Pro přípravu vzorků pro Western blot jsou využívány lyzační pufry, které uvolní a rozpustí proteiny obsažené v testovaných buňkách a tkáních.
Při výběru vhodného lyzačního pufru se především řídíme požadavky použité protilátky. Protilátky obvykle rozeznávají na proteinu pouze jeho malou část (označovanou jako epitop), ke které se váží. Tato část ovšem může být schovaná terciární strukturou proteinu, proto je třeba tuto strukturu proteinu nejprve rozplést, tj. protein denaturovat. Většina protilátek tedy rozeznává proteiny v jejich denaturované formě, nicméně jsou protilátky, které naopak vyžadují protein v nativní formě a protein nerozeznají, pokud byl připraven v roztoku obsahujícím některý z detergentů (SDS, deoxycholát). Tyto protilátky totiž rozeznávají své epitopy v závislosti na sekvencích aminokyselin, které se na základě trojrozměrné struktury proteinu nacházejí v blízkosti epitopu. Před započetím Western blotu je proto zcela zásadní zkontrolovat požadavky dané protilátky, a to v materiálech dodaných od výrobce.
Pokud protilátka vyžaduje nativní formu proteinu, je třeba protein připravit v pufru bez detergentu, případně v pufru, který obsahuje některý z mírně působících detergentů jako je např. Triton X-100 nebo NP-40.
Při výběru lyzačního roztoku se rovněž řídíme lokalizací testovaného proteinu, viz tabulka.
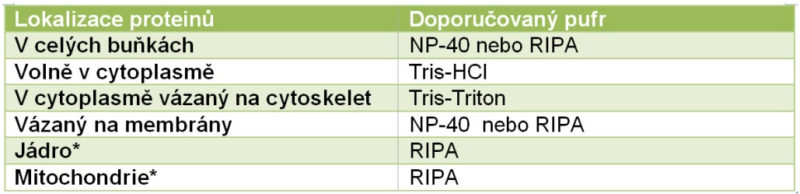
* Zejména u proteinů, u nichž předpokládáme nízkou expresi a které se nachází pouze v jádru nebo pouze v mitochondriích, se doporučuje připravit lyzát pouze z těchto buněčných částí.
Nonidet-P40 (NP40) pufr: Je vhodným pufrem pro studium proteinů nacházejících se v cytoplasmě, vázaných na membránách nebo proteinů z extraktů celých jader.
RIPA pufr(Radio Immuno Precipitation Assay buffer): Je používaným pufrem pro extrakty z celých buněk a proteiny vázané na membrány. Protože obsahuje iontové detergenty, které snadno vedou k rozpuštění proteinů v roztoku, je vhodným pufrem v případě špatně rozpustných tkání. Nicméně také porušuje protein-proteinové interakce, takže není vhodným roztokem při imunoprecipitačních experimentech.
Tris-Triton pufr: Je vhodným pufrem pro extrakci cytoskeletálních proteinů.
Tris- HCl pufr: Je vhodným pufrem pro použití s protilátkami, které vyžadují proteiny v jejich nedenaturované, tj. nativní formě, protože pro minimalizaci denaturace je třeba využít pufry, které neobsahují iontové detergenty (tj. SDS) a ideálně také neiontové detergenty (tj. Triton – 100).
Inhibitory proteáz a fosfatáz. Při přípravě lyzátu mohou v lyzované tkáni nastat nežádoucí rozkladné procesy. Při přípravě proteinových lyzátů jsou nežádoucí zejména aktivity proteáz a fosfatáz. Proto pro minimalizaci jejich aktivity je doporučováno do lyzačního roztoku, těsně před jeho použitím, přidat inhibitory proteáz a fosfatáz. Vhodné koktejly inhibitorů lze zakoupit komerčně.
Příprava lyzátu. Pro další minimalizaci aktivity proteáz a fosfatáz je třeba vzorky připravovat v předem vychlazeném lyzačním roztoku (na 4°C) a vzorky držet při 4°C i nadále. Při přípravě tkáně (např. její pitvě) postupujeme rychle, aby se snížilo riziko rozkladných procesů v tkáni. Pro přípravu homogenátu jsou využívány homogenizátory, homogenizaci můžeme zefektivnit tím, že tkáň předem zmrazíme, rozmělníme a urychleně přidáme vychlazený lyzační pufr. Množství přidaného pufru je úměrné množství tkáně a množství tkáně by optimálně mělo být 1-5 mg na 1 ml lyzačního roztoku. Vzorky jsou inkubovány 2 hod. při 4°C. Po následné centrifugaci je odebrán supernatant a přenesen do čisté zkumavky.
————————————————————————————————————————————————————————————–
URČENÍ KONCENTRACE CELKOVÝCH PROTEINŮ
Připravený lyzát je třeba vyhodnotit na koncentraci proteinů. Pro vyhodnocení koncentrace proteinů je obvykle využívaná Bradfordova nebo BCA metoda. Pro tyto metody existuje celá řada komerčních produktů. Po vyhodnocení koncentrace může být část vzorku nanesena na gel, případně může být vzorek v alikvotech zamražen při -20°C nebo -80°C pro pozdější použití.
————————————————————————————————————————————————————————————–
PŘÍPRAVA VZORKŮ PRO NANESENÍ NA GEL
Vzorky v denaturované a redukované formě
Jak již bylo zmíněno, většina protilátek vyžaduje proteiny v jejich denaturované formě. Proto ve většině případů musíme proteiny denaturovat. Denaturace proteinů je docíleno jednak působením neiontového detergentu SDS (dodaný součástí nanášecího, tzv. Laemmliho pufru) a také zahřátím směsi na 95-100°C po dobu 5 min.
2X Laemmliho pufr (4% SDS, 10% 2-merkaptoetanol, 20% glycerol, 0.02% bromfenolová modř, 120mM Tris-HCl, pH 6,8). Laemmliho pufr je běžně používaný nanášecí denaturační pufr.
• Součástí pufru je SDS, které jednak zajišťuje denaturaci proteinů, ale také zajistí, že proteiny budou mít negativní náboj (proteiny se totiž vážou na anionty SDS). Díky tomu se proteiny v gelu nepohybují na základě svého elektrického náboje, ale na základě své molekulární velikosti.
• ß-merkaptoetanol, případně ditiotreitol (DTT) jsou využity k odstranění intramolekulárních a intermolekulárních disulfidických můstků v proteinu, a tedy k „narovnání“ molekuly proteinu.
• Glycerol zvýší densitu nanášeného vzorku a usnadní tak jeho nanesení do jamky.
• V nanášecím roztoku je přítomná také barvička, typicky bromfenolová modř, která umožní vizualizaci vzorku při nanášení a v průběhu elektroforetické separace.
Vzorky v nativní a ne-redukované formě
![]() Některé protilátky rozeznávají své epitopy v závislosti na sekvencích aminokyselin, které se na základě troj-rozměrné struktury proteinu nacházejí v blízkosti epitopu. V těchto případech je nepřípustné připravovat vzorek za přítomnosti denaturačních látek (jako SDS) a také je nepřípustné zahřívání vzorku na denaturační teplotu. Rovněž některé protilátky protein rozpoznají, jen pokud je ve své ne-redukované formě. V těchto případech nanášecí a elektroforetický pufr nesmí obsahovat redukční látky jako je ß-merkaptoetanol a DTT. V těchto případech je využívaný 2X nanášecí pufr (60 mM Tris-HCl, pH 6.8, 20% glycerol, 0,02% bromfenolové modře).
Některé protilátky rozeznávají své epitopy v závislosti na sekvencích aminokyselin, které se na základě troj-rozměrné struktury proteinu nacházejí v blízkosti epitopu. V těchto případech je nepřípustné připravovat vzorek za přítomnosti denaturačních látek (jako SDS) a také je nepřípustné zahřívání vzorku na denaturační teplotu. Rovněž některé protilátky protein rozpoznají, jen pokud je ve své ne-redukované formě. V těchto případech nanášecí a elektroforetický pufr nesmí obsahovat redukční látky jako je ß-merkaptoetanol a DTT. V těchto případech je využívaný 2X nanášecí pufr (60 mM Tris-HCl, pH 6.8, 20% glycerol, 0,02% bromfenolové modře).
————————————————————————————————————————————————————————————–
ELEKTROFORETICKÁ SEPARACE PROTEINŮ
Pro separaci proteinů je využívaná PAGE elektroforéza, tj. elektroforéza v polyakrylamidovém gelu.
Polyakrylamidové gely. Polyakrylamid (poly(2-propenamid)) je polymer tvořený z akrylamidových podjednotek zesíťovaných N,N‘-methylenbisakrylamidem (zkráceně bisakrylamidem). Směs akrylamidu a bisakrylamidu polymerizuje při pokojové teplotě pomocí volných radikálů, které dodává persulfát amonným (APS) způsobující homolytické štěpení vazeb O-O. K urychlení polymerizace se používá volná zásada TEMED (tetrametylendiamin), který katalyzuje tvorbu volných radikálů persulfátu amonného. Při práci s akrylamidem je třeba obezřetnosti, protože působí jako neurotoxin.
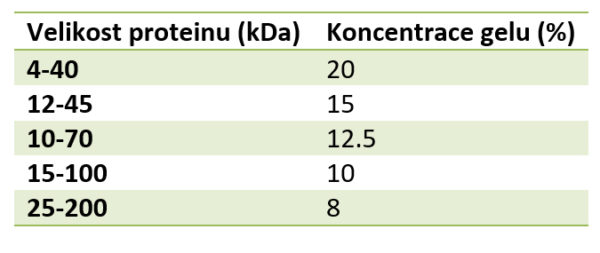
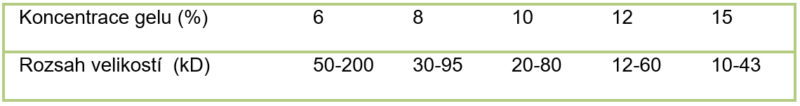
Při přípravě polyakrylamidového gelu lze výběrem vhodné koncentrace akrylamidu a bisakrylamidu ovlivňovat velikost pórů gelu a tím optimální separaci proteinů o určité velikosti. Se zvyšující se koncentrací těchto látek se póry gelu stávají menšími a gel se hodí pro separaci menších molekul, viz tabulka. Gely mohou být zakoupeny komerčně nebo se mohou připravit v laboratoři.
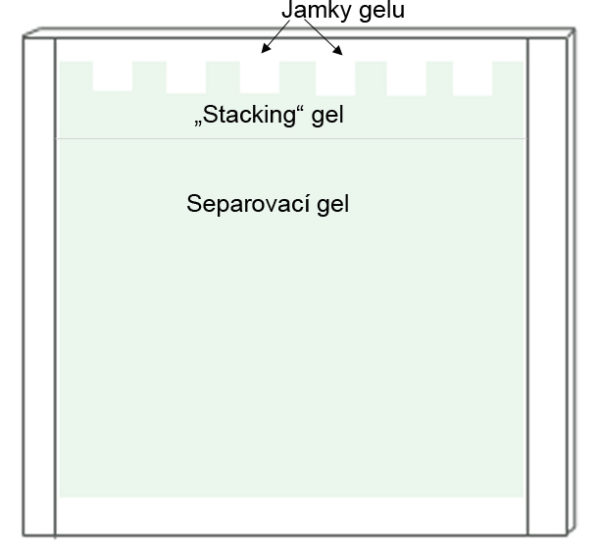
„Stacking“ a separovací gel. Při přípravě gelu pro separaci proteinů se velmi často využívá kombinace dvou gelů. Gel je tvořen horním úsekem (blíže ke startu migrace) tzv. „stacking“ gelem o nízké koncentraci akrylamidu a nízkém pH (6,8), v úseku dále od startu je tzv. separovací gel o vyšším pH (8,8) a vyšší koncentraci akrylamidu. Stacking gel svou nízkou koncentrací umožní vysokomolekulárním molekulám snadnější „rozjezd“ a tím jim zabrání, aby svou velikostí zablokovaly póry gelu a znesnadnily tak vstupu a rozjezdu nízkomolekulárních molekul.
SDS-PAGE. Pokud separujeme proteiny v denaturované formě (nejčastěji), využíváme techniku tzv. SDS-PAGE (Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis), tj. připravujeme gel přídavkem SDS. Působením SDS dochází jednak k denaturaci proteinů a také proteiny získávají negativní náboj, a to tím, že se navazují na anionty SDS. Díky tomu se pohybují od kladného pólu k zápornému a rychlost jejich migrace závisí na jejich molekulové hmotnosti. Pomocí SDS-PAGE jsou tedy proteiny separovány na základě své molekulové hmotnosti.
Nativní-PAGE.Pokud separujeme proteiny v jejich nativní formě (méně často), využíváme techniku tzv. Nativní-PAGE (gel připravujeme bez SDS). Při Nativní PAGE jsou proteiny separovány na základě svého celkového náboje a své hydrodynamické velikosti. Náboj proteinu závisí na skladbě aminokyselin (poměrem kyselých a zásaditých aminokyselin) a také post-translačních modifikací. Většina proteinů má izoelektrický bod v oblasti kyselé či mírně zásadité a migruje směrem k zápornému pólu.
![]() Akrylamid je neurotoxin. Práce s ním vyžaduje obezřetnost.
Akrylamid je neurotoxin. Práce s ním vyžaduje obezřetnost.
————————————————————————————————————————————————————————————–
NANÁŠENÍ VZORKŮ
Množství vzorků, které se nanáší na jednu jamku (mini-gelu) by mělo obsahovat 20-40 μg celkových proteinů. Pro snadnější nanášení se vzorky nanáší speciálními, dlouhými a tenkými špičkami.
————————————————————————————————————————————————————————————–
ELEKTROFORETICKÝ PUFR
Jako elektroforetický pufr se standardně využívá 1X Tris-glycin-SDS pufr (25 mM Tris base 190 mM glycin, 0.1% SDS, pH 8.3) při SDS-PAGE a 1X Tris-glycin pufr (25 mM Tris, 190 mM glycin) při nativní-PAGE.
————————————————————————————————————————————————————————————–
PROTEINOVÝ MARKER A NANÁŠECÍ KONTROLY
Proteinový marker. Do jedné z jamek je rovněž třeba nanést proteinový marker, což je komerčně dostupná směs proteinů o známé velikosti. Proteinový marker nám slouží pro vyhodnocení velikosti signálů v testovaných vzorcích.
Nanášecí kontroly. V případech, kdy protein potřebujeme kvantifikovat, je doporučováno využití také protilátky proti strukturálnímu proteinu (jako je GAPDH, aktin nebo tubulin). Exprese strukturálních proteinů by měla být u všech testovaných vzorků stejná, tzn. intenzita výsledného signálu pro tyto proteiny by po imunodetekci měla být také stejná. Pokud by byly jednotlivé vzorky naneseny v rozdílném množství, nebo by došlo k nerovnoměrnému přenosu z gelu na membránu, můžeme za použití strukturálních proteinů provést normalizaci.
————————————————————————————————————————————————————————————–
ELEKTROFORETICKÝ PŘENOS A PŘENOS VZORKŮ Z GELU NA MEMBRÁNU
Při elektroforéze dbáme podmínek stanovené výrobcem elektroforetické aparatury.
Pro přenos proteinů z gelu na membránu se v současné době nejvíce využívá elektroforetického transferu, tzv. elektroblotu, tj. působení elektrického proudu. Jako membrány jsou využívány nitrocelulosové nebo častěji polyvinylové (PVDF). Při přenosu dbáme podmínek stanovené výrobcem aparatury.
————————————————————————————————————————————————————————————–
ZVIDITELNĚNÍ CELKOVÝCH PROTEINŮ
Vizualizace celkových proteinů lze dosáhnout několika způsoby. Jedním z nich je barvení proteinů pomocí Coomassie blue (0.25% Coomassie Brilliant Blue R-250 v 10% kyselině octové / 50% metanolu). Pomocí Coomassie blue barvíme proteiny přímo v gelu po jejich elektroforetické separaci. Citlivost barvení pomocí Coomassie blue není nijak velká, detekční limit je 0,3 – 1 ug proteinu v proužku. Barvení pomocí Coomassie blue ukazuje úspěšnost separace a stejně tak abundanci celkových proteinů v jednotlivých vzorcích. Coomassie blue zůstává navázaná na proteinech.
Druhým častým způsobem je barvení pomocí Ponceau S neboli Ponceau S red (0.1% (w/v) Ponceau S v 5% kyselině octové). Barvení pomocí Ponceau S je až po transferu proteinů na membránu. Barvíme tedy proteiny na membráně. Barvení pomocí Ponceau S je reverzibilní a je odstraněno při promývacích krocích. Vizualizací celkových proteinů pomocí Ponceau S jednak vyhodnotíme účinnost transferu a jednak podobně jako s Coomassie blue získáme informaci o celkovém množství nanesených proteinů v jednotlivých vzorcích a poloze proužků proteinového markerů. Citlivost barvení s Ponceau S je asi 2x silnější než s Coomassie blue.
————————————————————————————————————————————————————————————–
BLOKOVÁNÍ NESPECIFICKÉHO VÁZÁNÍ PROTILÁTEK NA MEMBRÁNU
Membrány, které jsou využívány pro Western blot mají obvykle vysokou afinitu k proteinům, tedy i k protilátkám. Proto je třeba zajistit to, aby se protilátky při inkubaci s membránou navazovaly specificky ke svým antigenům na vzorcích a ne celkově k povrchu membrány. Provádí se proto tzv. blokování membrány v blokovacích roztocích, které zaplní volná místa na membráně a protilátka se váže pouze na svůj specifický antigen. Existuje široká škála blokovacích roztoků, od těch, které obsahují sušené odtučněné mléko či BSA (bovinní sérum albumin) k roztokům s vysoce purifikovanými proteiny. Účinnost blokování může záviset na použité protilátce, tzn. v některých případech je třeba vhodnost blokovacího roztoku zjistit empiricky. Nejčastěji jsou ovšem používány blokovací roztoky 5% sušeného mléka či lépe 5% BSA. Obecně ovšem platí, že využití mléka není vhodné při detekci fosforylovaných proteinů. Blokování membrány v blokovacím roztoku probíhá obvykle 1 hod při 4°C za konstantního, lehkého protřepávání.
————————————————————————————————————————————————————————————–
PROMÝVACÍ ROZTOKY
V průběhu procedury je membrána s navázanými proteiny promývaná v promývacích roztocích. Promývání membrány zajistí odstranění či snížení pozadí, které je způsobené nespecifickým navázáním protilátek a zajistí tak čistotu signálu. Nedostatečné promývání má za následek zvýšení nechtěného pozadí, naopak příliš intenzivní promývání vede ke ztrátě citlivosti detekce.
Jako promývací roztoky jsou nejčastěji využívány TBS (Tris buffered saline) nebo PBS (phosphate buffered saline) s přídavkem detergentů (pro snížení nespecifického pozadí) jako je například 0,05% Tween 20. Koncentrace detergentu může být ovšem různá v závislosti na individuálním experimentu, a může se pohybovat od 0,05% do 0,5%.Typicky, promývací roztoky obsahují i blokovací roztok (v ředění 1:10). Zahrnutí blokovacího roztoku do promývacího roztoku vede k zabránění vyvazování blokovacího proteinu z membrány do promývacího roztoku, což opět vede k minimalizaci nespecifického pozadí.
Co se týče přípravy promývacích roztoků s detergenty a zvlášť s těmi obsahujícími blokovací roztok, je doporučována příprava vždy čerstvých roztoků před samotnými experimenty. Skladováním těchto roztoků narůstá nebezpečí vzniku mikrobiální kontaminace, která může výrazným způsobem přispět k nespecifickému pozadí.
————————————————————————————————————————————————————————————–
PROTILÁTKY
• Příslušný protein navázaný na membráně je detekován pomocí primární protilátky.
Výběr primární protilátky je vázán příslušným antigenem a komerční dostupností dané protilátky.
• Protože primární protilátky nelze obvykle přímo vizualizovat, je třeba pro vizualizaci použít značenou sekundární protilátku, která rozpoznává primární protilátku a váže se na ni. Sekundární protilátka je označena nějakou značkou (např. biotinem) nebo konjugována s nějakým reportérovým enzymem umožňující vizualizaci – např. s peroxidázou (HRP, z angl. horseradish peroxidase) či alkalickou fosfatázou (AP, z angl. alkaline phosphatase).
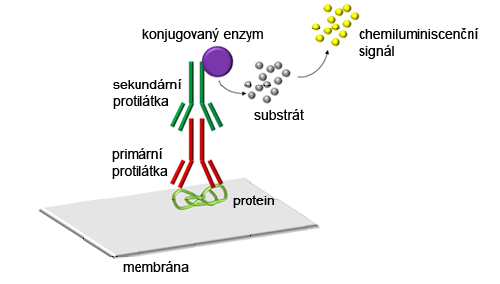
• Komerčně je dostupná celá škála sekundárních protilátek, nicméně při výběru sekundární protilátky musíme respektovat hostitelský organismus, ve kterém byla připravena primární protilátka. Takže např. pokud je primární protilátka připravena v myši, sekundární protilátka musí být proti myši (anti-mouse) a získaná v jiném hostitelském organismu než je myš.
Inkubace s primární protilátkou
Primární protilátky jsou pro experimenty ředěny, a to nejčastěji v rozsahu 1/100 – 1/500 000 ze zásobního roztoku 1mg/ml. Optimální ředění pro daný experiment je nutné zjistit empiricky. Inkubace s primární protilátkou je obvykle v blokovacím roztoku, méně častěji v PBS nebo TBS s přídavkem 0,1% Tween 20 (roztoky PBST a TBST), případně v PBST nebo TBST s nízkou koncentrací sušeného mléka nebo BSA (0,5-0,25%). Vhodnost použití blokovacího roztoku nebo PBST či TBST je vhodné otestovat pro danou protilátku. Je třeba si uvědomit, že přítomnost blokovacího agens jednak vede ke snížení nespecifického pozadí, ale na druhou stranu sníží i specifický signál.
Inkubační čas je závislý na vazebné schopnosti protilátky k proteinu a také množství proteinu. Obvykle se inkubační doba s primární protilátkou pohybuje od několika málo hodin až do 18 hodin. Inkubace probíhá obvykle při 4°C za konstantního lehkého protřepávání. Při déle trvající inkubaci je teplota 4°C podmínkou pro zamezení mikrobiální kontaminace a tím degradaci proteinů.
Inkubace se sekundární protilátkou
Sekundární protilátka je obvykle naředěna v promývacím roztoku (ředění sekundární protilátky přímo v blokovacím roztoku sice snižuje nespecifické pozadí, ale také snižuje intenzitu specifického signálu). Vhodné ředění je obvykle udáno v manuálu od výrobce a obvykle se pohybuje v rozsahu od 1:1000 – 1:20 000. Inkubační doba je 1-2 hodiny při 4°C za konstantního promíchávání. Pro optimální výsledek je obvykle zjistit vhodné ředění empiricky. Příliš velké ředění vede k nízkému signálu, naopak příliš nízké ředění vede k velkému nespecifickému pozadí.
————————————————————————————————————————————————————————————–
DETEKČNÍ SYSTÉMY
V minulosti se pro detekci hojně využívaly radioizotopy, nicméně v současné době již využívány nejsou a detekce je založena nejčastěji na enzymatických reakcích a také fluorescenci. Pro enzymatickou detekci je nejčastěji využíván systém na základě alkalické fosfatázy (alkaline phosphatase, AP) nebo křenové peroxidázy (horseradish peroxidase, HRP). Hojně je využívaný chemiluminiscenční systém, kdy enzym reaguje s dodaným chemiluminiscenčním substrátem a přeměňuje jej na nestabilní produkt, který se stabilizuje vyzářením kvanta světla a vzniká tak chemiluminiscenční signál. Množství signálu je poté úměrné množství navázaného enzymu, které je přímo úměrné množství detekovaného proteinu. Uvolňované světlo je pak detekováno použitím CCD-kamerou. Obdobně je využívaná kolorimetrická reakce.
————————————————————————————————————————————————————————————–
PROTOKOL
PŘÍPRAVA ELEKTROFORETICKÉ APARATURY
Saponátem a vodou se důkladně umyjí skla, plastové vložky a hřebínek, poté se důkladně opláchnou deionizovanou/destilovanou vodou a etanolem a nechají se uschnout.
![]() Se skly se manipuluje jen v rukavicích, přičemž se skla drží za jejich okraje. Skla nesmí být jakkoliv znečištěna, aby bylo zabráněno tvorbě bublin při nalévání gelu.
Se skly se manipuluje jen v rukavicích, přičemž se skla drží za jejich okraje. Skla nesmí být jakkoliv znečištěna, aby bylo zabráněno tvorbě bublin při nalévání gelu.
Skla se sestaví k sobě, vloží boční vložky, okraje skel se mohou olepit izolepou pro ujištění, že nedojte k vytečení roztoku gelu. Skla se zajistí svorkami a vloží se vertikálně do připravené aparatury.
Dle velikosti separovaných molekul připravíme separovací gel s vhodnou koncentrací (viz tabulka). Jednotlivé komponenty přidáváme v uvedeném pořadí, viz. tabulka. Po přídavku TEMEDu, roztok ihned rychle promícháme a vlijeme mezi připravená skla. Horní okraj gelu by měl být 1 cm od spodního okraje hřebínku. Gel převrstvíme vrstvou vody nebo isopropanolu a necháme polymerizovat ve vertikální poloze asi 30 min.
Přídavkem TEMEDu akrylamid začíná rychle polymerizovat.
Převrstvení gelu brání difúzi kyslíku do gelu. Kyslík by v horní části gelu potlačoval polymerizaci.
Tabulka. Koncentrace gelu dle velikosti separovaného proteinu.
Tabulka. Separovací gel (20 ml). Připravujte v uvedeném pořadí.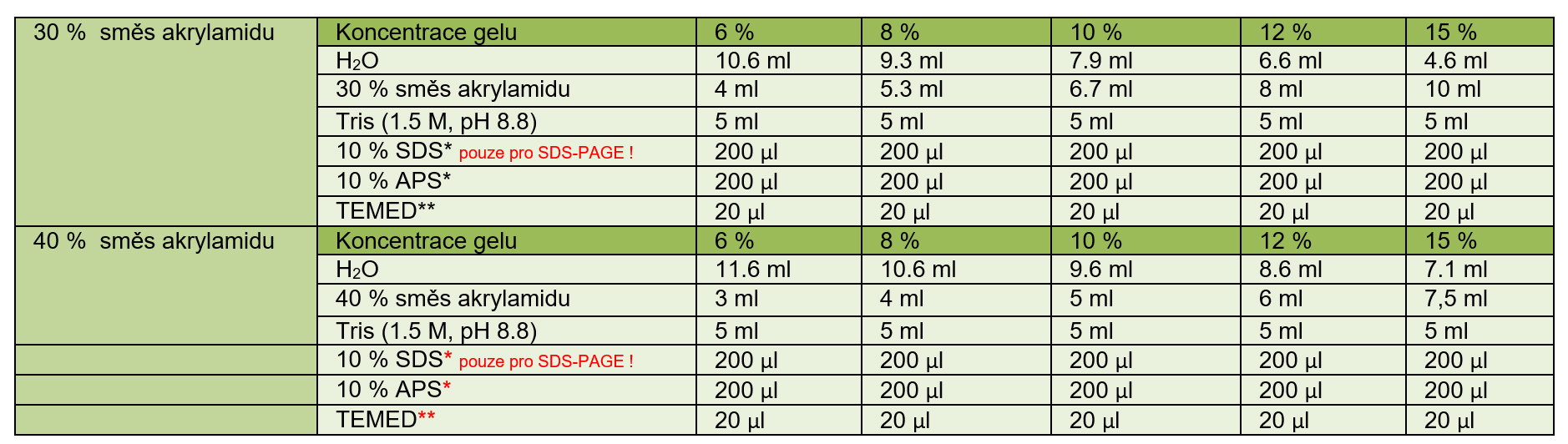
* před přidáním komponenty roztok řádně promíchejte
** řádně promíchejte a ihned vlijte mezi připravená skla a převrstvěte izopropanolem
Z připraveného gelu odstraníme vrstvu izopropanolu či vody. Z povrchu gelu odstraníme nezpolymerizovaný akrylamid několikerým propláchnutím vodou. Vodu odstraníme a povrch gelu dokonale vysušíme vložením filtračního papíru.
Připravíme „stacking“ gel (viz tabulka). Jednotlivé komponenty přidáváme v uvedeném pořadí. Po přidání TEMEDu, roztok rychle zamícháme a vlijeme mezi skla k připravenému separačnímu gelu. Vložíme hřebínek (dbáme na to, abychom do gelu nevtlačili bubliny) a necháme polymerovat cca 30 min.
Tabulka. „Stacking“ gel (typicky 5%). Připravujte v uvedeném pořadí.
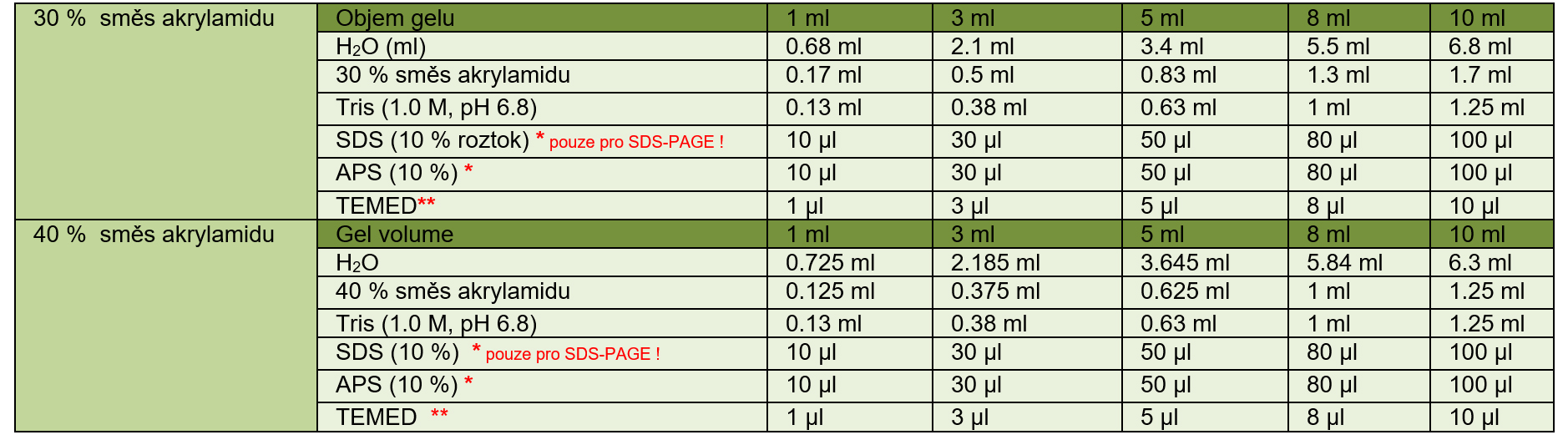 * před přidáním komponenty roztok řádně promíchejte
* před přidáním komponenty roztok řádně promíchejte
** řádně promíchejte a ihned vlijte mezi připravená skla a vložte hřeben
————————————————————————————————————————————————————————————–
PŘÍPRAVA VZORKU
Testovaný vzorek i proteinový marker smícháme s nanášecím pufrem tak, aby celkový objem vzorku byl 10-15 μl. Testovaný vzorek by měl obsahovat 20-40 μg celkových proteinů na jamku.
Pokud provádíme SDS-PAGE, tj. proteiny denaturujeme, smícháme vzorky i marker v poměru 1:1 s Laemmliho pufrem (4% SDS, 10% 2-merkaptoetanol, 20% glycerol, 0.004% bromfenolová modř, pH 6.8). Vzorky i marker zahřejeme na 100°C po dobu 3 min.
Pokud testované proteiny jsou extrémně hydrofobní, např. proteiny obsahující mnohonásobné transmembránové domény, mohou tyto proteiny při zahřívání precipitovat. Aby se této precipitaci zamezilo, je doporučeno denaturaci provádět při teplotě 45-50°C po dobu 1 hod.
Pokud provádíme Nativní-PAGE, smícháme vzorky v poměru 1:1 s 2X nanášecím pufrem (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 1% Bromfenolové modře).
————————————————————————————————————————————————————————————–
NANESENÍ VZORKU NA GEL A ELEKTROFORETICKÁ SEPARACE
• Z gelu odstraníme hřebínek. Z jamek důkladně odmyjeme veškerý nezpolymerizovaný akrylamid, a to buď proudem vody pomocí kádinky, nebo promýváním pipetou v jednotlivých jamkách.
Nepropláchnutí jamek způsobí nerovnou separaci proteinů v dané jamce a tím křivé výsledné proužky po imunodetekci.
• Připravený gel vložíme do elektroforetické aparatury. Do horního a spodního zásobníku nalijeme elektroforetický pufr:
SDS-PAGE: 1X Tris-glycin-SDS pufr (25 mM Tris base 190 mM glycin, 0.1% SDS, pH 8.3)
Nativní-PAGE: 1X Tris-glycin pufr (25 mM Tris, 190 mM glycin)
• Připojíme elektroforetickou aparaturu ke zdroji napětí tak, že záporný pól je nahoře a kladný dole. Aplikujeme napětí o 8 V/cm mezi elektrodami. Ve chvíli, kdy vzorky (bromfenolová modř) postoupí z „stacking“ pufru do separačního, zvýšíme napětí na 15 V/cm.
• Elektroforézu ukončíme ve chvíli, kdy se bromfenolová modř dostane ke spodnímu okraji gelu. Rozebereme elektroforetickou aparaturu, rozebereme skla s gelem a gel překlopíme na filtrační papír. Pro pozdější orientaci gelu, jeden roh gelu odstřihneme.
————————————————————————————————————————————————————————————–
BARVENÍ POMOCÍ COOMASSIE BLUE
• Nejprve inkubujeme gel ve fixačním roztoku (50% metanol, 10% ledová kyselina octová) po dobu 1 hodiny až přes noc, za mírného protřepávání. Po první hodině vyměníme fixační roztok za čerstvý.
• Gel barvíme v barvícím roztoku (0,1% Coomassie Brilliant Blue R-250, 50% metanol, 10% ledová kyselina octová), za mírného protřepávání. Barvíme tak dlouho, až gel získá barvu barvícího roztoku.
• Gel inkubujeme v odbarvovacím roztoku (40% metanol, 10% ledová kyselina octová) tak dlouho, dokud není pozadí na gelu plně odbarveno a proužky nejsou jasně viditelné.
• Gel můžeme uchovat ve skladovacím roztoku (5% ledová kyselina octová).
————————————————————————————————————————————————————————————–
PŘENOS NA MEMBRÁNU
• Připravíme 1x Electrotransfer Buffer (100 ml 10x Electrotransfer Buffer, 200 ml metanol, 700 ml dH2O).
– 10x Electrotransfer Buffer (30.3 g Tris Base, 144 g Glycin, doplnit do 1 l)
• Připravíme přenosovou aparaturu dle instrukcí výrobce.
• Připravíme PVDF membránu: membránu namočíme do 100% methanolu na 20 sec, poté vložíme do dH2O na 2 min a poté do 1x Transfer pufru na 5 min.
• Provedeme transfer, obvykle při 35-40 V přes noc. Přenosovou aparaturu chladíme, např. pomocí chladícího gelu či ji provádíme v chladné místnosti.
————————————————————————————————————————————————————————————–
BARVENÍ POMOCÍ PONCEAU-S
• Membránu ponoříme do roztoku Ponceau-S (0,1% Ponceau S, 1% kyselina octová) a necháme barvit po dobu 5 min.
• Membránu odbarvíme použitím odbarvovacího roztoku(1% kyselina octová po dobu 5 min) tak, aby se odbarvilo pozadí a vyjevily se proužky proteinů.
————————————————————————————————————————————————————————————–
BLOKOVÁNÍ MEMBRÁNY A INKUBACE S PRIMÁRNÍ PROTILÁTKOU
• Membránu inkubujeme 1-3 hodiny v blokovacím roztoku (1x PBS, 5% sušeného, odtučněného mléka, 0.05% Tween 20).
Membrána nesmí uschnout!!!
• Membránu lze s blokovacím roztokem a následně i s protilátkou inkubovat v plastovém sáčku se zavařenými či zalepenými okraji
• Promyjeme 3 x 5 min v 1x PBS, 0.05% Tween 20.
• Inkubujeme s primární protilátkou (ředění 1 : 1000 v 1x PBS, 5% sušeného mléka,+ 0.05% Tween 20. Inkubujeme 1 hodinu při 4 °C).
————————————————————————————————————————————————————————————–
PROMYTÍ MEMBRÁNY A INKUBACE SE SEKUNDÁRNÍ PROTILÁTKOU
• Promyjeme 3 x 5 min v 1x PBS + 0.05% Tween 20 za pokojové teploty.
• Inkubujeme v 1x PBS + 10% sušeného mléka + 0.05% Tween 20 po dobu 10 min.
• Inkubujeme se sekundární protilátkou (ředění 1 : 5000), po dobu 30 min za pokojové teploty v 1x PBS, 5% sušeného mléka, 0.05% Tween 20).
• Promyjeme 3 x 5 min v 1x PBS + 0.05% Tween 20 za pokojové teploty.
————————————————————————————————————————————————————————————–
PROMYTÍ MEMBRÁNY A INKUBACE SE SEKUNDÁRNÍ PROTILÁTKOU
Detekujeme sekundární protilátku s použitím příslušného systému.